Introducing Evo Protein System 0.0.I

Table of Contents
- Introduction Evo Protein System 0.0.I
- Features and Capabilities
- Standard Version (Coming Soon)
- Prompting Examples
- Enterprise Version (Limited Access)
- Distribution and Access
Introduction Evo Protein System 0.0.I
When we started Frontier Evo at the beginning of November 2024, we positioned ourselves with the belief that integrating simulation capabilities into AI is essential for developing frontier systems that can assist humans and further accelerate scientific and engineering progress on a broad scale. Recognizing the potential of AI to revolutionize various fields, we set out with a clear vision to harness its power for meaningful advancements.
Today, we are excited to announce a closed research preview of our Evo Protein System (v0.0.1), which will be fully integrated into our Evo Cloud Platform alongside Cibo-1. This release represents our initial research into augmenting protein science workloads with AI, leveraging state-of-the-art biological foundation models (bFMs), advanced techniques, and computational models. As part of our Cibo-1 research initiative, the Evo Protein System will enhance the efficiency and accuracy of protein science workflows, potentially enabling breakthroughs in medicine, biotechnology, and related domains.
This closed preview marks an important milestone in our journey toward developing human-in-the-loop language agents with physics-aware simulators and sophisticated subroutines. The Evo Protein System integrates large language models, interactive agents, and advanced protein science capabilities within a unified interface.
Features and Capabilities

Our system provides comprehensive capabilities, and almost every component has been written to support our project's objective—building All-In-One interface between language models and protein workflow capabilities.
Standard Version (Coming Soon)
While this release is currently limited to our research partners, we plan to make the standard version available more broadly in the future. Here are the key features that will be included:
- Automated protein motif scaffolding and predicting protein sequence,
- Hardware-aware (CPU/GPU) protein folding (Omegafold),
- Automated protein function prediction,
- Secondary structure editing (like alpha helices),
- Unbounded force energy calculation from sequences,
- Protein dynamics computations such as anisotropic network and Gaussian network models,
- Search and retrieval capabilities, tools for accessing protein information,
- In-depth analysis of protein structures from the Protein Data Bank (PDB),
- Customizable 3D protein structure visualizers, Web UI chat interface
Here are early showings from our system:
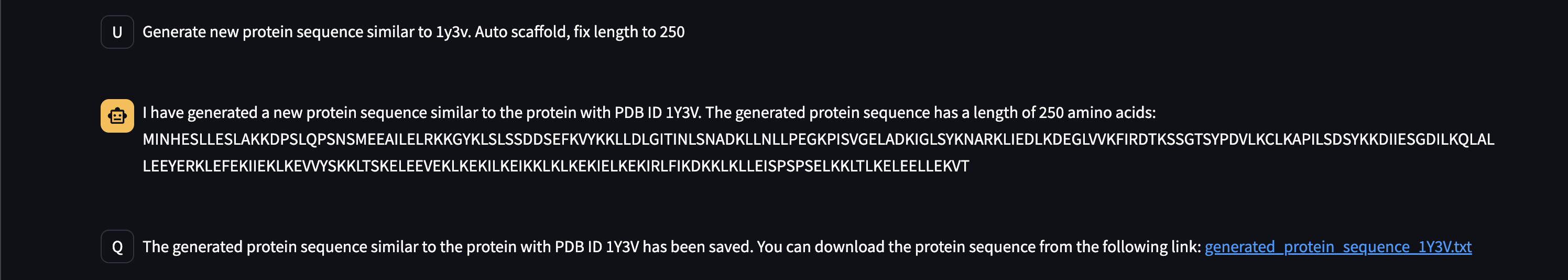
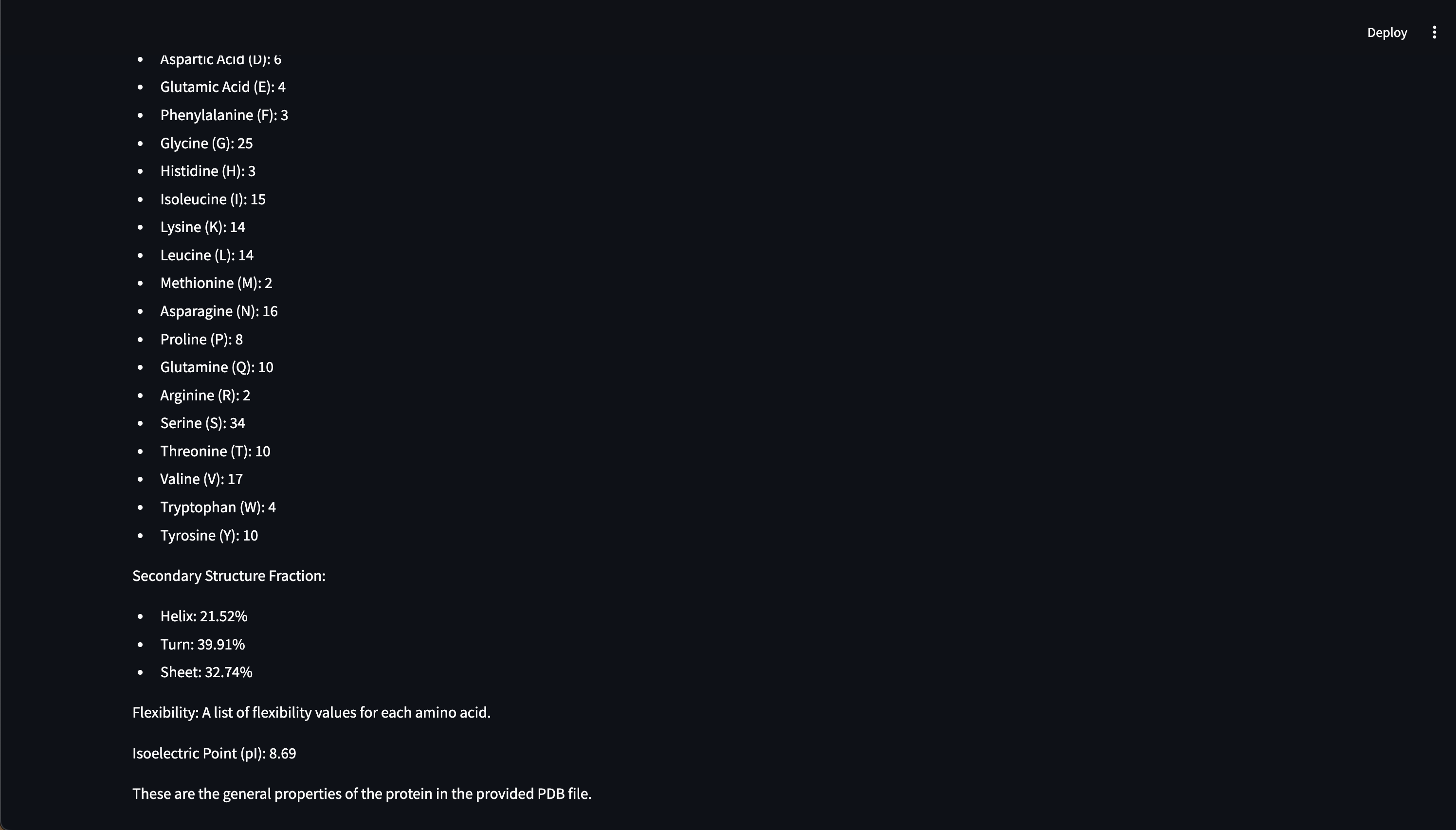
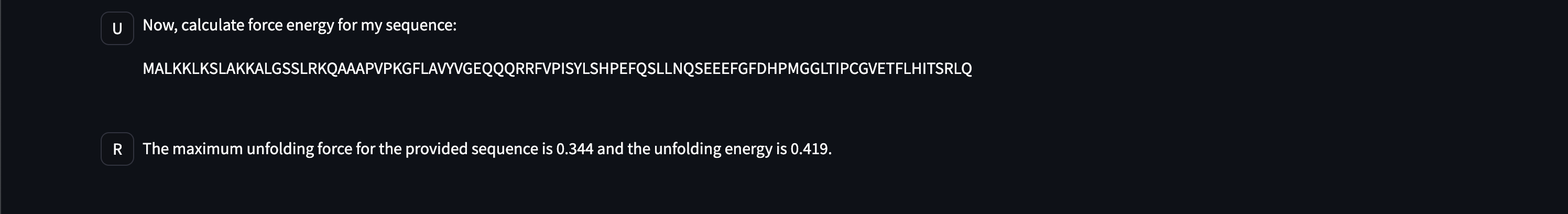
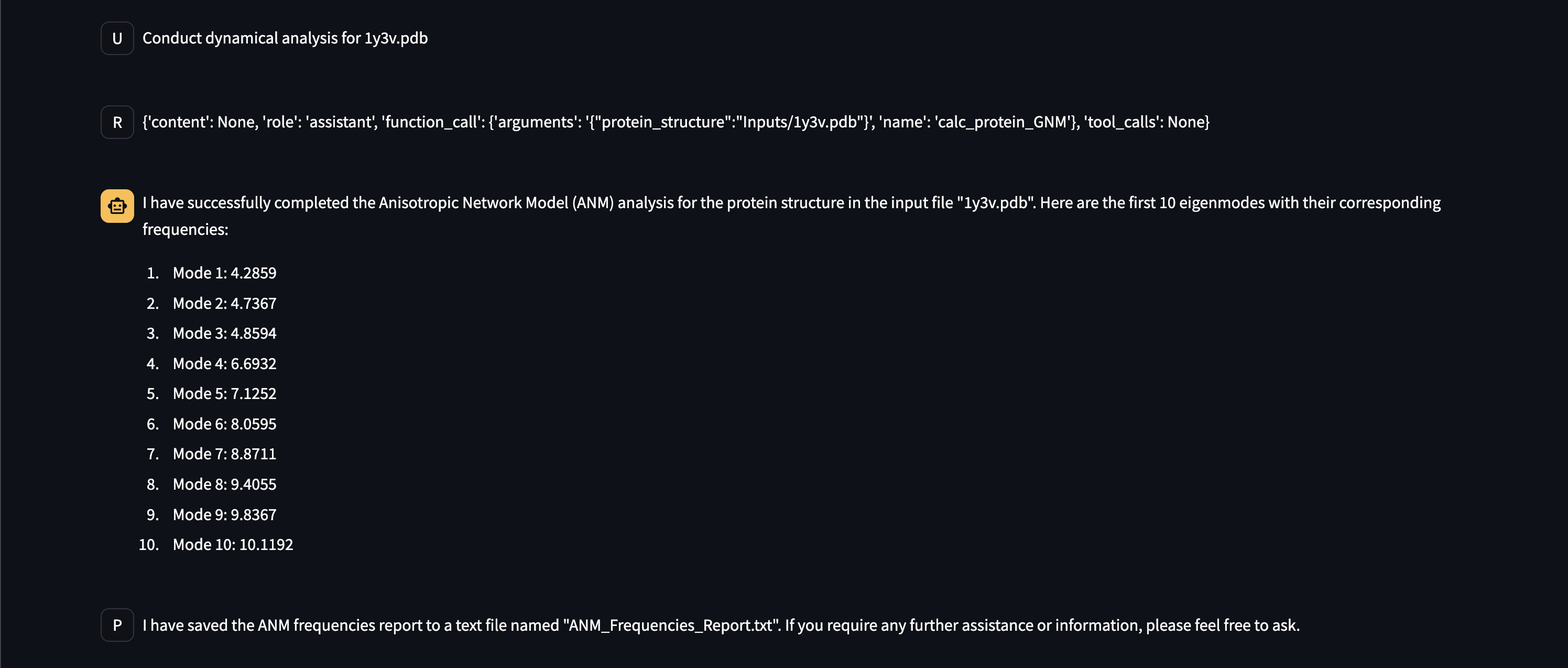
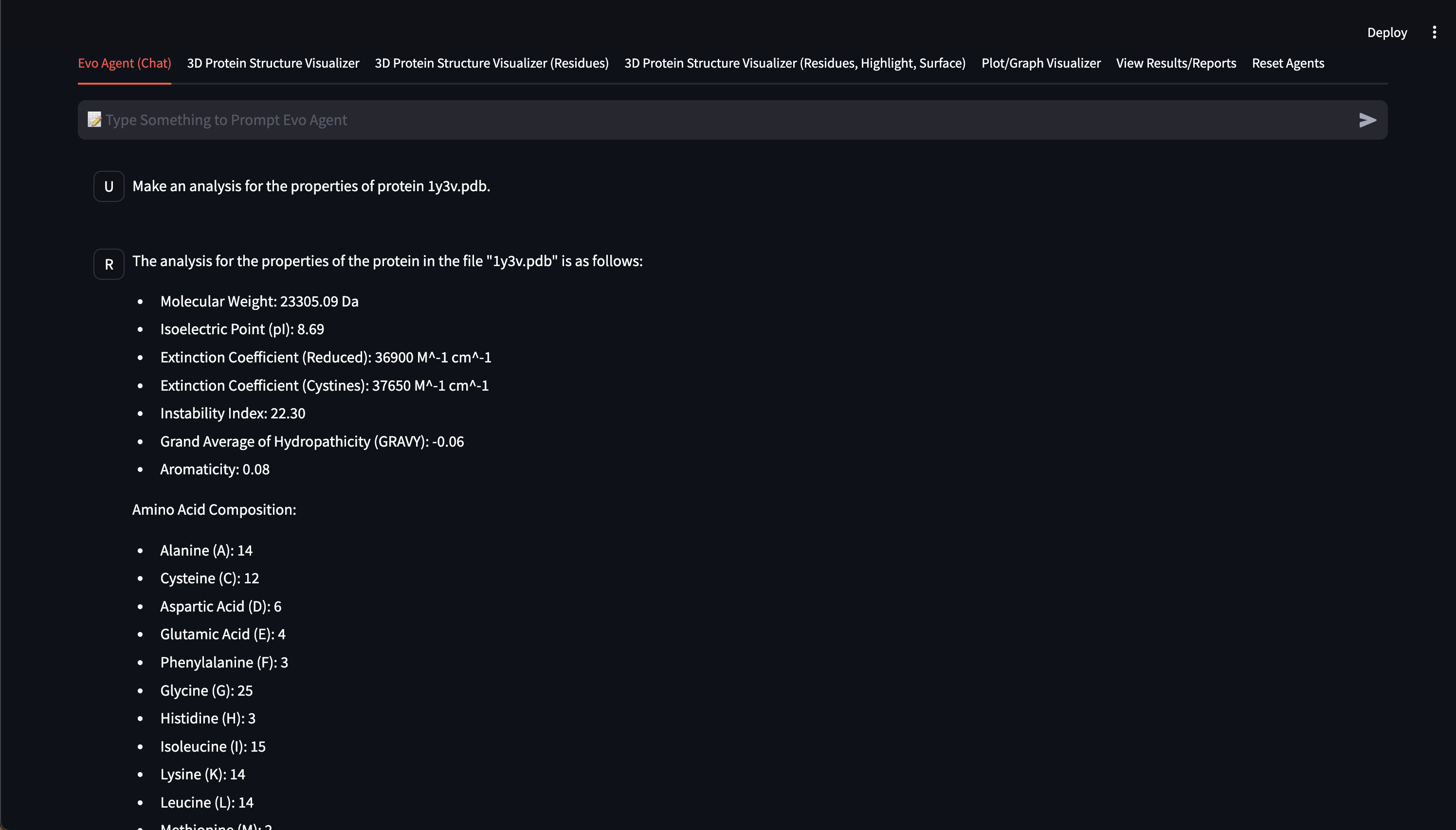
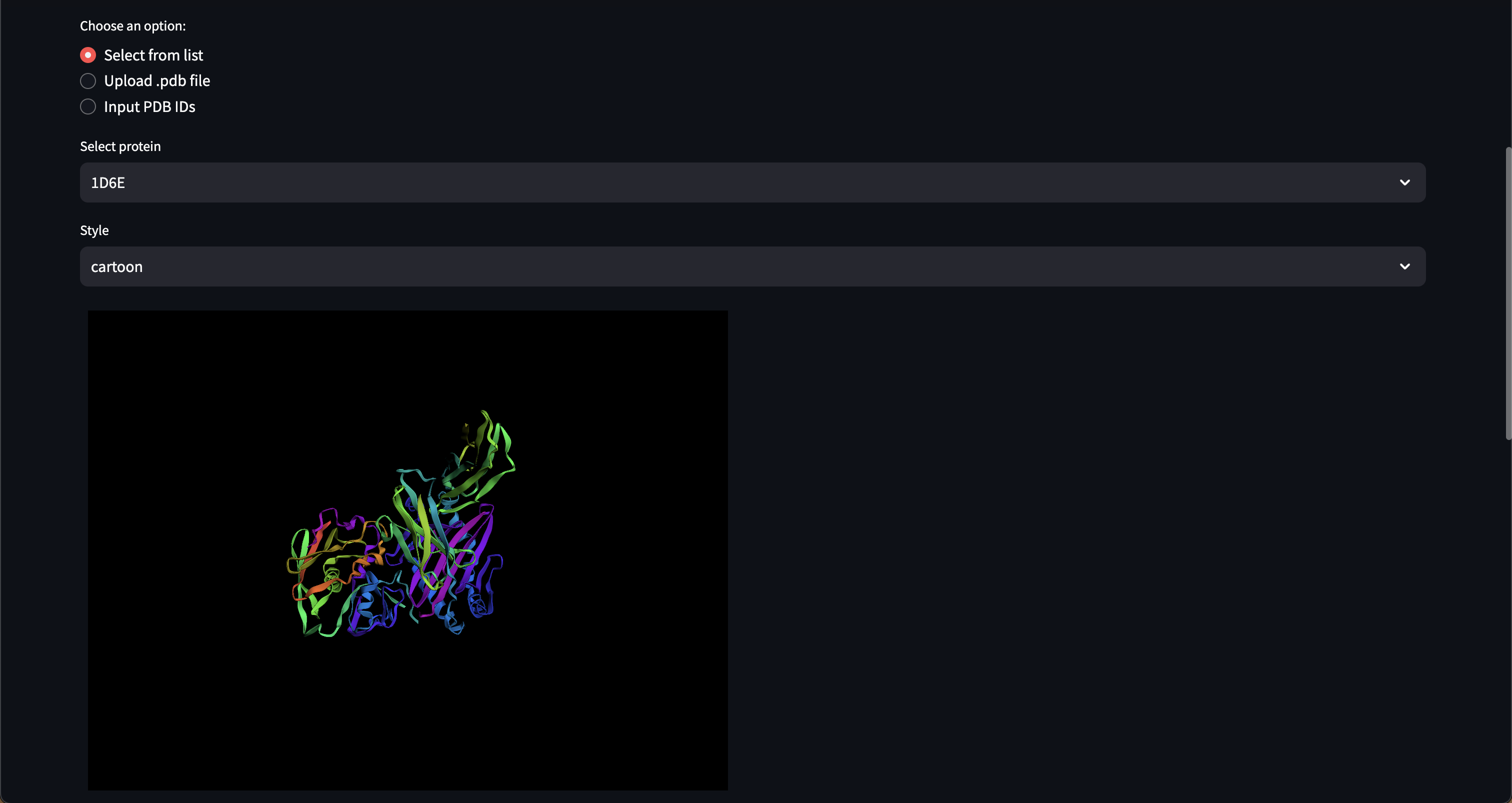
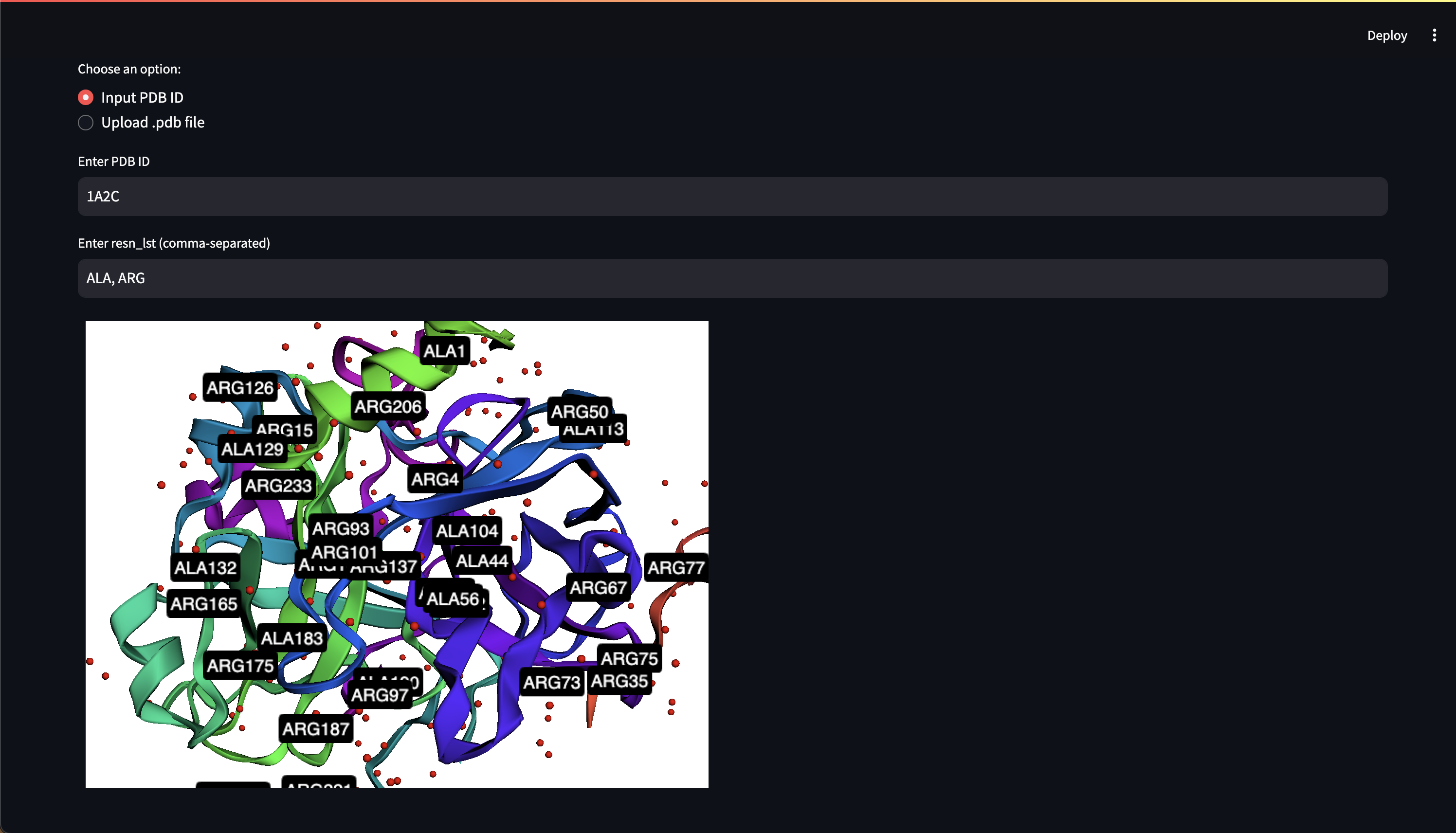
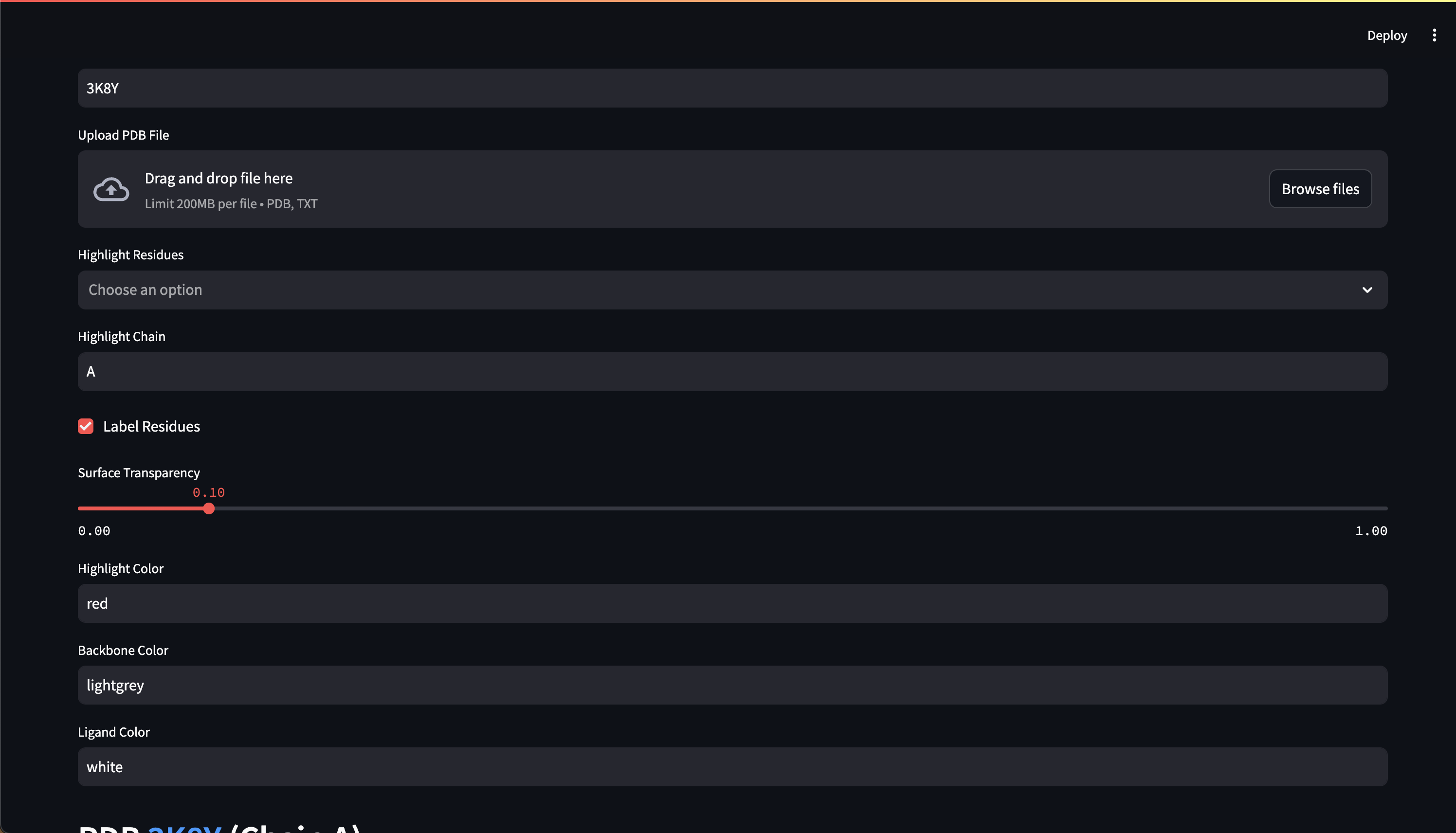
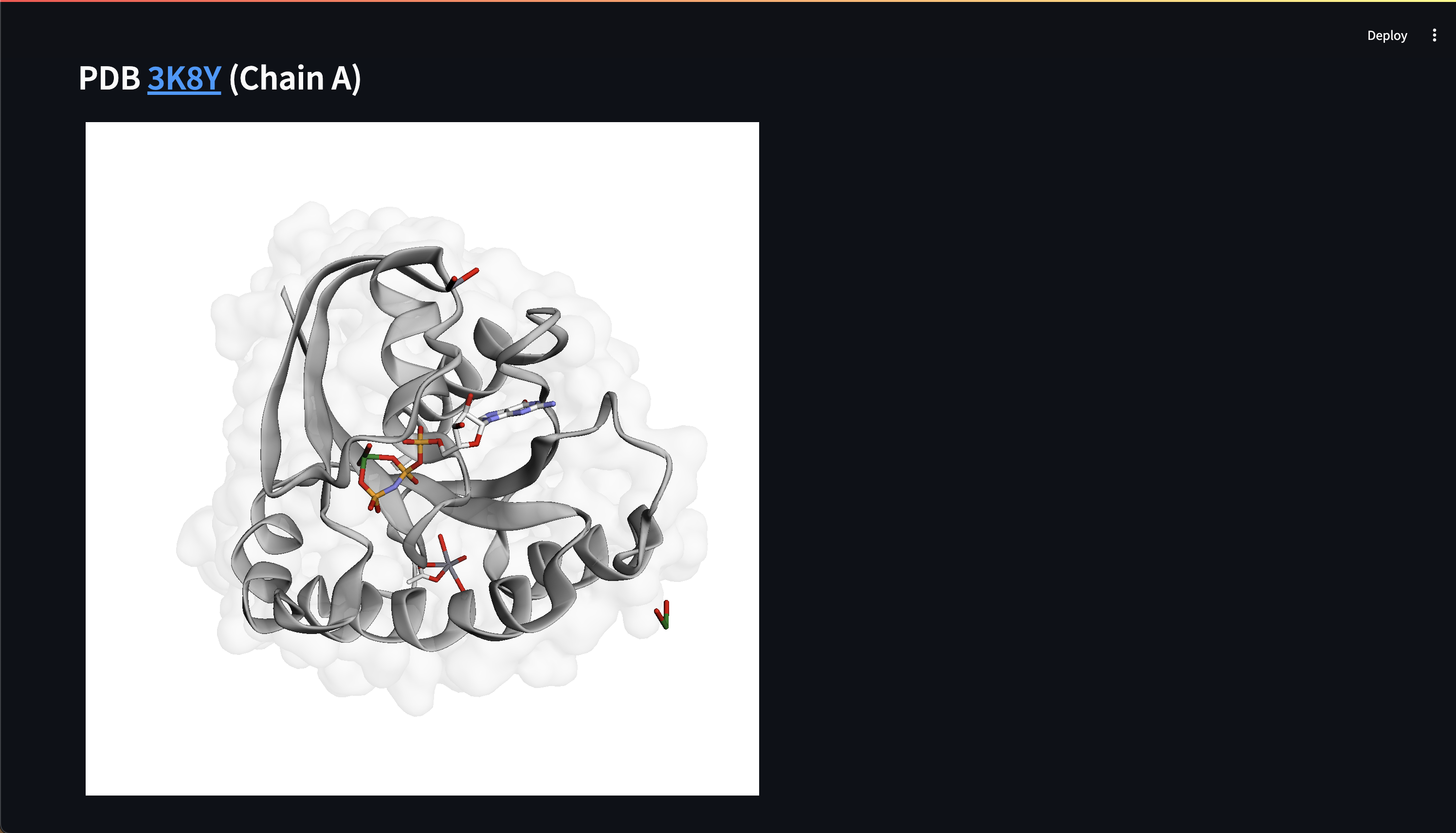
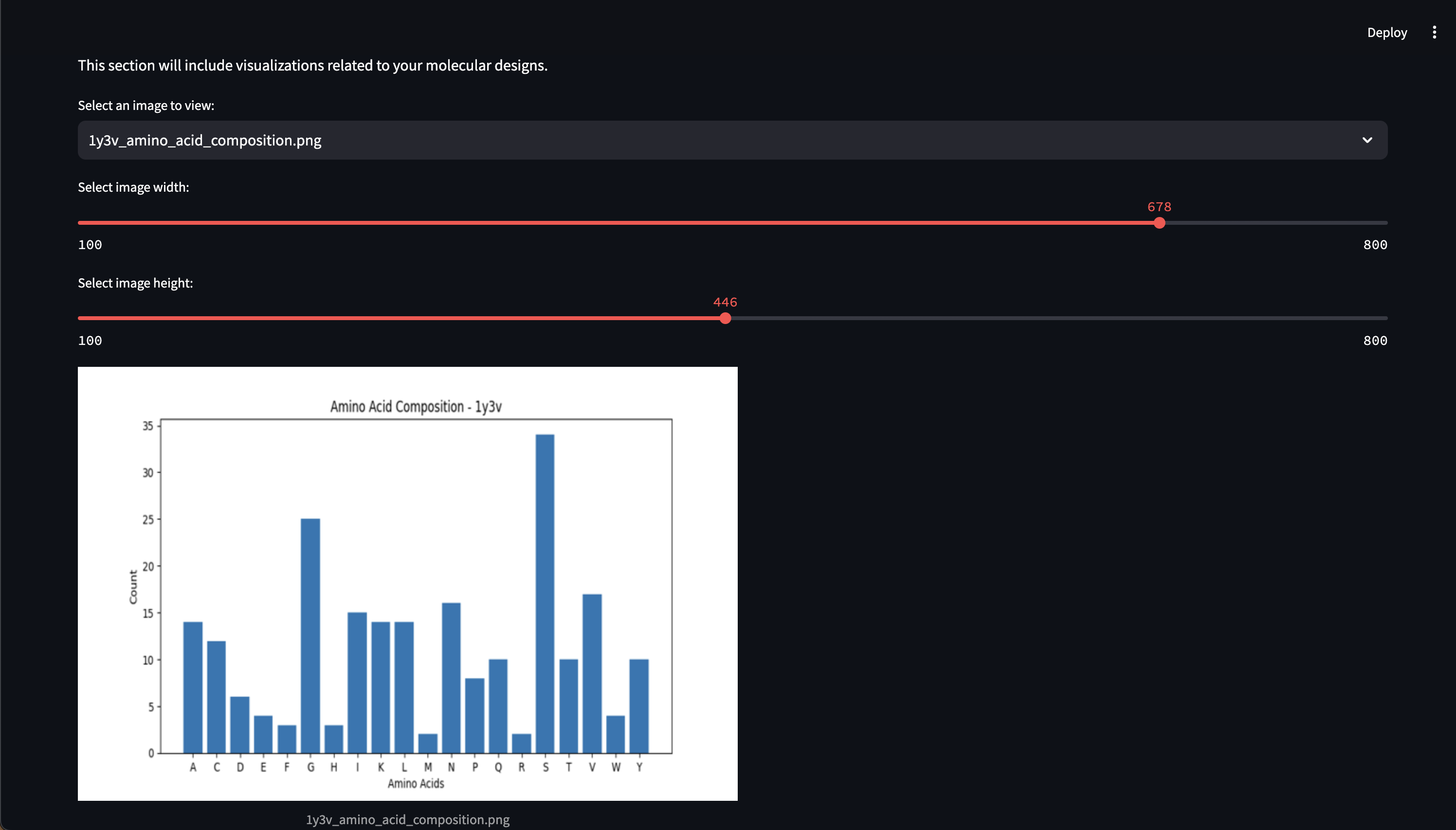
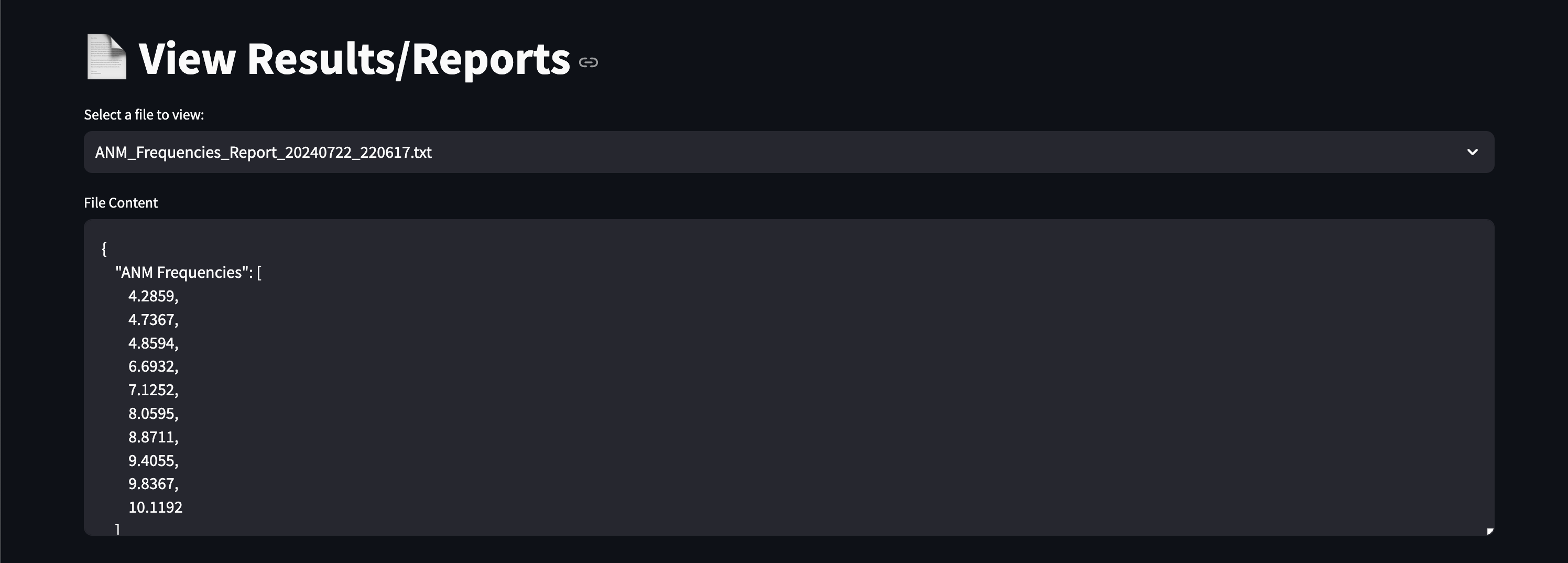
Prompting Examples
| Human Instruction | Planning | Output |
|---|---|---|
Analyze the functional domains present in the following protein sequence: MGDVEKGKKIFIMKCSQCHTVEKGGKHKTGPNLHGLFGRKTGQAPGYSYTAANKNKGIIWGEDTLMEYLENPKKYIPGTK | 1. Identify Functional Domains: - Run Pfam/SMART domain prediction - Analyze conserved motifs - Map secondary structure elements - Identify key residues 2. Sequence Alignment: - BLAST against UniProt database - Multiple sequence alignment - Phylogenetic analysis - Conservation scoring 3. Domain Analysis: - Structure-function relationships - Cofactor binding sites - Post-translational modifications - Interaction interfaces | 1. Domain Characterization: - Cytochrome C domain (residues 11-103): • Heme-binding motif: CXXCH at position 14-18 • Fe coordination: His18, Met80 • Redox-active residues: Cys14, Cys17 2. Evolutionary Analysis: - 98.2% identity to human cytochrome c - Conserved across eukaryotes (E-value: 1e-85) - Key mutations: None in catalytic site - Conservation score: 0.92 (highly conserved) 3. Functional Properties: - Redox potential: +260mV vs. NHE - Electron transfer rate: ~10⁶ s⁻¹ - Optimal pH range: 6.0-8.5 - Critical in respiratory chain between Complex III/IV |
| Human Instruction | Planning | Output |
|---|---|---|
| Design a protein sequence with enhanced thermal stability for industrial applications, focusing on the catalytic domain of a serine protease. | 1. Structural Analysis: - Map backbone hydrogen bond network - Calculate Rosetta energy terms - Analyze B-factors and flexibility - Identify destabilizing regions 2. Computational Design: - Run RosettaDesign simulations - Optimize core packing density - Calculate electrostatic interactions - Model conformational entropy 3. Validation Protocol: - Molecular dynamics at elevated temps - Free energy perturbation analysis - Active site geometry verification - Substrate docking simulations | 1. Design Optimizations: - Core Mutations: • V53I, A112L: Improved packing (ΔΔG = -1.8 kcal/mol) • G96P: Reduced backbone flexibility • N156D/K158R: New salt bridge network - Surface Engineering: • Added 3 surface ion pairs (E23R/D28K) • Helix dipole optimization (T45E) • Pro residues in loops (S76P, G89P) 2. Stability Metrics: - Tm increased by 12.3°C (from 65°C to 77.3°C) - ΔΔG = -4.2 kcal/mol at 25°C - t1/2 at 70°C: 4.8 hours (5x improvement) - Maintains activity up to 75°C 3. Functional Validation: - Catalytic triad geometry preserved (RMSD 0.3Å) - kcat/KM = 5.2 × 10⁵ M⁻¹s⁻¹ (85% of WT) - pH stability range expanded (5.5-9.0) - Retains 92% activity in 2M GuHCl |
| Human Instruction | Planning | Output |
|---|---|---|
Predict potential protein-protein interaction sites for the following sequence: MAEGEITTFTALTEKFNLPPGNYKKPKLLYCSNGGHFLRILPDGTVDGTRDRSDQHIQLQLSAESVGEVYIKSTETGQYLAMDTDGLLYGSQTPNEECLFLERLEENHYNTYISKKHAEKNWFVGLKKNGSCKRGPRTHYGQKAILFLPLPV | 1. Surface Analysis: - Calculate SASA (Solvent Accessible Surface Area) - Evaluate evolutionary conservation scores - Map electrostatic potential surface 2. Motif Detection: - Screen against PROSITE database - Analyze binding pocket geometry - Evaluate sequence entropy 3. Structure Prediction: - Generate AlphaFold2 model - Perform molecular dynamics simulation - Calculate binding energy landscape | 1. Interface Analysis: - Primary Interface (residues 71-85): • ΔG binding = -8.3 kcal/mol • Conservation score: 0.85 • Hydrophobic patch with φ/ψ angles in favorable regions • Key residues: Y73, F77, L82 form hydrophobic core - Secondary Interface (residues 120-134): • ΔG binding = -5.1 kcal/mol • Polar residues: D121, R127, K132 • Dynamic loop region (RMSF: 2.3Å) 2. Structural Characterization: - FGF binding motif (13-20): TTFTALT - Canonical β-trefoil topology (RMSD: 0.8Å) - Binding pocket volume: 1,243ų 3. Interaction Potential: - Predicted Kd: ~50nM for primary interface - pH-dependent binding (optimal pH 7.2-7.8) - Temperature stability up to 42°C |
These examples demonstrate the system's capability to:
- Perform detailed sequence analysis
- Design proteins with specific properties
- Predict protein-protein interactions
- Integrate multiple analytical approaches
- Provide actionable insights for protein engineering
Enterprise Version (Limited Access)
Our enterprise offering delivers advanced capabilities designed for cutting-edge research environments. Available exclusively to select research partners, it extends beyond standard features to enable sophisticated protein analysis at scale.
Key enterprise capabilities include:
-
Complete Standard Feature Set
- Full access to all standard version capabilities
- Priority compute resources and support
-
Advanced Multi-Agent System
- Sophisticated multi-agent reasoning framework
- Seamless integration of diverse biological datasets
- Real-time multimodal processing (numerical, textual, spatial)
- Enhanced decision support through comprehensive data analysis
-
Advanced Protein Analysis Suite
- State-of-the-art protein-protein complex clustering
- Persistent homology alignment for structural analysis
- Topological feature detection and mapping
- High-precision sequence alignment algorithms
- Advanced structural relationship modeling
These enterprise capabilities enable organizations to push the boundaries of protein research while maintaining the highest standards of precision and reliability. Our system's sophisticated analysis tools, combined with multi-agent reasoning, provide unprecedented insights into protein structures and interactions.
Distribution and Access
The Evo Protein System is currently available only through our closed research preview program and will be integrated into the Evo Cloud Platform by the end of Q4 2024. Working in conjunction with Cibo-1, our Level 2 autonomous research system, it will provide researchers with a comprehensive suite of tools for protein science and biological discovery. While we plan to release the standard version more broadly in the future, access is currently limited to research partners and select enterprise clients. If you're interested in participating in our research program or discussing enterprise access, please contact us to learn more about potential collaboration opportunities.